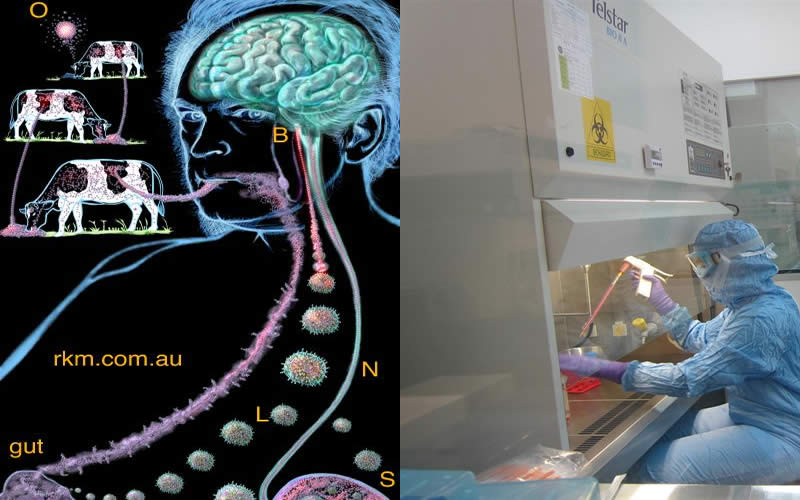

MADRID (España).- Las proteínas son parte de la extraordinaria maquinaria que es el cuerpo humano y cuando funcionan mal provocan enfermedades. Este es el caso de los priones, proteínas que son infecciosas cuando están mal plegadas y que están detrás de la enfermedad de Creutzfeldt-Jakob o el insomnio familiar letal.
En los últimos años, la comunidad científica ha ido dando pasos para comprender su complejo funcionamiento y ha establecido que, al igual que en los virus, existen distintas cepas en los priones que son las que provocan el tipo y/o grado de gravedad de la enfermedad.
En sentido estricto, el prion es solo aquella proteína mal plegada que causa enfermedades priónicas, como la de Creutzfeldt-Jakob (en ganado esta se llama mal de las vacas locas).
Sin embargo, ahora se empieza a hablar de proteínas tipo prion para referirse a una proteína mal plegada que se propaga induciendo sobre otras este mal plegamiento, como las que existen detrás de enfermedades neurodegenerativas como el alzhéimer o párkinson.
En un caso y en otro se produce un mal funcionamiento de la proteína -proteína beta-amiloide en caso de alzhéimer- y en los dos -enfermedades neurodegenerativas y priónicas- parecen existir cepas que «definen» el tipo de patología y/o sintomatologías diferentes.
¿Pero cómo funcionan? ¿Por qué algunas cepas de priones se acumulan en el tálamo provocando una enfermedad concreta y otras lo hacen, por ejemplo, en la corteza cerebral ocasionando otra forma de enfermedad? Un nuevo estudio liderado por españoles y publicado en la revista Acta Neuropathologica intenta ahondar en este puzzle.
El trabajo lo dirige Joaquín Castilla, científico Ikerbasque (Fundación Vasca para la Ciencia) en el Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), con sede en Bilbao (norte), y según relata a Efe, este abre la puerta a nuevas dianas para crear futuros tratamientos de patologías priónicas o neurodegenerativas.
La investigación se ha hecho ‘in vitro’ y es resultado de experimentos realizados en los últimos cinco años.
La mayor dificultad ha sido la de poder distinguir las distintas cepas ya que, aunque tienen propiedades biológicas singulares, son muy similares; conseguido esto, los investigadores trataron de averiguar cómo estas se dirigen a una zona u otra del cerebro.
Y es que los científicos ya sabían que las cepas priónicas son, en definitiva, las responsables de una u otra patología, pero sospechaban, además, que tales diferencias no podían deberse únicamente a las proteínas mal plegadas.
Así, encontraron, y esto es lo que se describe en este artículo, que existen distintos cofactores -moléculas en las que se repiten subunidades de carga negativa- que ayudan a estas proteínas mal plegadas a depositarse en una u otra zona del cerebro (las que se agregan en el tálamo provocan, por ejemplo, insomnio familiar fatal y las que lo hacen en la corteza cerebral, Creutzfeldt-Jakob).
Estos cofactores explicarían las distintas localizaciones de las cepas en el sistema nervioso central y suponen un factor más para comprender el desarrollo de muchas enfermedades y para, en un futuro, generar nuevos tratamientos o mejorarlos, concluye Castilla.
La investigación se ha desarrollado en colaboración con la Universidad de Santiago de Compostela, la de Salamanca, el Laboratorio Central de Veterinaria de Madrid y el Instituto Superior de Sanidad de Italia.
Los priones son partículas no celulares, son proteínas que sin ser virus, tienen también características patógenas e infecciosas.
⊕